

Souhrn
Působením ATM kinázy dochází k fosforylaci a vzestupu množství proteinu p53, který působí jako transkripční faktor a má v regulaci buněčného cyklu a indukci apoptózy zásadní význam. Mezenchymální kmenové buňky jsou nehematopoetické kmenové buňky přítomné v kostní dřeni a schopné diferenciace do různých typů buněčných linií. Mezenchymální kmenové buňky s podobnou biologickou charakteristikou jako buňky z kostní dřeně byly izolovány také z dalších zdrojů, jako je např. zubní pulpa. Velkou výhodou právě kmenových buněk zubní pulpy je nejen jejich snadná získatelnost, ale i vysoká interaktivita s biomateriály, což z nich činí možné ideální elementy pro rekonstrukci tkání. Otázkou zůstává, jakým způsobem tyto buňky reagují na genotoxický stres a jím vyvolané poškození DNA. Zdá se, že poškozené buňky neumírají apoptózou jako v případě kmenových buněk krvetvorby, ale pouze ztrácí schopnost proliferace, akumulují se v G1 a G2 bloku buněčného cyklu. Tento stav se nazývá senescence.
Summary
Ćmielová, J., Muthná, D., Vávrová, J., Řezáčová, M. Genotoxic stress and DNA damage in stem cells
Genotoxic stress causes cell cycle arrest and damage reparation or apoptosis induction. One of the particularly important form of DNA damage represent double strand breaks. ATM (ataxia telangiectasia mutated) kinase activation is the first step in cellular response to this type of DNA damage. ATM kinase induces phosphorylation and increase in protein p53, a transcription factor which has an important role in cell cycle regulation and apoptosis induction. Mesenchymal stem cells are nonhematopoietic stem cells which occur in bone marrow and are able to differentiate into various types of tissue.
Mesenchymal stem cells have similar biological characteristic as bone marrow stem cells which were isolated also from other sources, for example dental pulp. Human dental pulp stem cells are an easily available source of human stem cells and their interactivity with biomaterials makes them ideal for tissue reconstruction. How these cells react to genotoxic stress and to DNA damage connected with it? Recently obtained informations show that damaged cells do not die by apoptosis in comparison with hematopoietic stem cells, but only lose their ability to proliferate and accumulate in G1 and G2 cell cycle arrest. This stage is called senescence.
Odpověď buněk na poškození DNA
DNA eukaryotických buněk je neustále vystavena působení genotoxického stresu, tedy různým endogenním a exogenním vlivům, jako jsou ionizující záření (IR), UV záření a chemické mutageny. Ty vyvolávají celou řadu poškození DNA, která vedou ke dvěma hlavním směrům biologické odpovědi v buňkách. Prvním je zástava buněčného cyklu v G1/S a G2/M kontrolních bodech, která umožňuje získání času pro reparaci poškození, druhým pak indukce buněčné smrti mechanismem apoptózy, pokud jsou poškození příliš rozsáhlá.(1) Jedno z nejzávažnějších poškození a velký zásah do integrity genomu představují DSB – dvojité zlomy DNA.(2)
DSB způsobují remodelaci chromatinu a tvorbu tzv. ionizujícím zářením indukovaných ohnisek (foci). Prvním krokem v odpovědi savčích buněk na DSB indukované ionizujícím zářením je aktivace ATM kinázy. Ozářením buněk dochází k její rychlé autofosforylaci na serinu 1981, což způsobí disociaci dimeru ATM a jeho aktivaci.(3) Aktivaci ATM podporuje komplex reparačních proteinů Mre11, Rad50, NBS1 (MRN komplex) a fosforylovaný histon H2AX (?-H2AX), které se hromadí v blízkosti poškození. Fosforylovaný H2AX tvoří ložiska v přítomnosti dvojitých zlomů DNA způsobených ionizujícím zářením a hraje důležitou roli v opravě DNA poškození.
Reakce, při níž dochází k fosforylaci H2AX, je velice rychlá, první fosforylované molekuly se objevují už ve 20 sekundách po ozáření a množství ?-H2AX se zvyšuje se vzrůstajícím množstvím poškození. Tuto fosforylaci mohou katalyzovat ATM, ATR (ataxia telangiectasia related) a DNA-dependentní protein kináza.(4) Aktivovaná ATM kináza řídí průchod buněčným cyklem, a to ovlivněním aktivity kontrolních kináz Chk1 (checkpoint kinase 1) a Chk2 (checkpoint kinase 2). V odpovědi na ionizující záření fosforyluje ATM kináza Chk1 na serinech 317 a 345. Aktivovaná Chk1 dále fosforyluje inaktivní fosfatázy Cdc25 (serine/threonine protein phosphatase), které hrají důležitou roli v regulaci buněčného cyklu.
Fosfatázy Cdc25 jsou schopné aktivovat nebo naopak inaktivovat cyklin dependentní kinázy Cdk1 (cyclin dependent kinase 1) a Cdk2 (cyclin dependent kinase 2), a to jejich fosforylací/defosforylací na tyrozinu 15 a treoninu 14. Defosforylace uvedených cyklin dependentních kináz vede k aktivaci, zatímco fosforylace vede k inaktivaci těchto komplexů. K defosforylaci, a tím pádem aktivaci Cdk1 může dojít také přes fosforylaci Cdc25 Chk1 kinázou na serinu 216. Fosforylací Cdc25 se tvoří vazebné místo pro proteiny rodiny 14–3–3, které umožňují transport Cdc25 do cytoplazmy a jeho následnou degradaci. Chk1 je tedy nezbytně nutná pro inhibici syntézy DNA indukovanou ionizujícím zářením a pro kontrolní bod G2/M, protože fosforyluje již zmíněné fosfatázy Cdc25, a tím nedochází k aktivační defosforylaci Cdk (Obr.).(5)
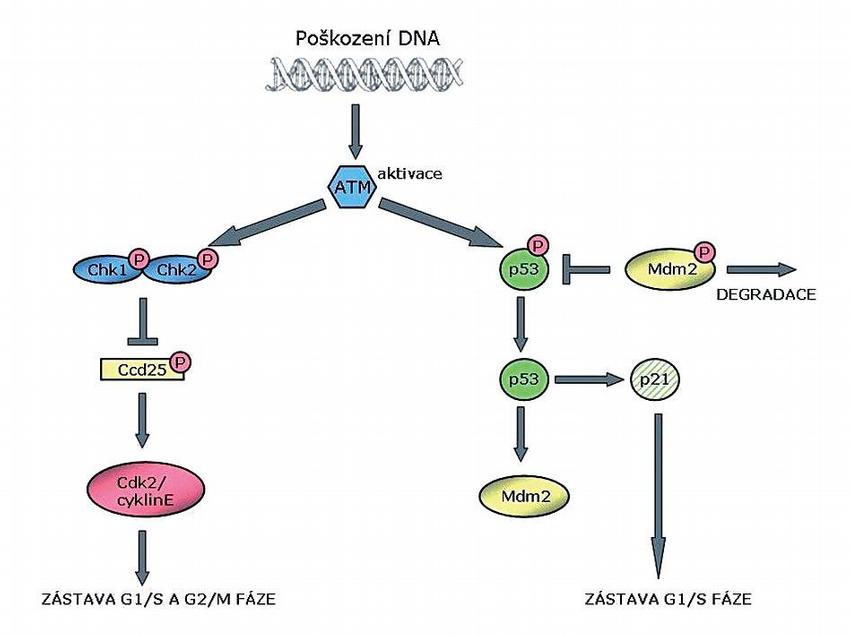
Dalším ze substrátů ATM je protein p53. Nádorový supresor p53 je protein, který odpovídá na různé typy buněčných stresorů tím, že indukuje zastavení buněčného cyklu, apoptózu, senescenci, opravu DNA nebo změnu metabolismu.(6) Aktivace proteinu p53 zahrnuje jeho stabilizaci, přeměnu latentní formy ve formu aktivní a umístění proteinu v jádře. V nestresovaných buňkách je protein p53 degradován pomocí Mdm2 (p53 binding protein) proteinu, což je považováno za nejdůležitější mechanismus regulace aktivity p53. V normálních buňkách se tedy protein p53 nachází v nízkých hladinách. Mdm2 působí jako ubikvitin ligáza a exportuje p53 z jádra buňky do cytoplazmy k degradaci v proteasomu.
Na poškození DNA navazuje vzestup množství p53 díky fosforylaci p53 v oblasti, kde se Mdm2 váže. Tato fosforylace se nejčastěji odehrává na serinu 15 působením kináz ATM a ATR. Kromě Mdm2 se na degradaci proteinu p53 v nestresovaných buňkách podílí také jun kináza (JNK). Zatímco se komplexy Mdm2 a proteinu p53 vyskytují specificky v S a G2/M fázi buněčného cyklu, JNK – p53 komplexy se přednostně nacházejí v G0/G1 fázi. Bylo zjištěno, že Mdm2 a JNK působí nezávisle na sobě.(7) Funkční protein p53 je významný transkripční faktor pro několik cílových genů, které jsou klíčové pro regulaci buněčného cyklu (zástava buněčného cyklu v G1/S kontrolním bodě skrze aktivaci proteinu p21), reparaci poškození genetického materiálu (protein Gadd 45 podněcuje reparaci DNA vyštěpením, a to buď přímo nebo v kooperaci s molekulami PCNA – proliferating cell nuclear antigen), navození apoptózy (významná úloha genu Bax).(8)
Protein p53 může být posttranslačně modifikován nejméně na 18 místech. V odpovědi na ionizující a UV záření v exponovaných buňkách je na N-konci fosforylováno 7 serinů a 2 treoniny (Ser 9, 15, 20, 33, 37, 46 a Thr 18 a 81). Thr 55 je fosforylován v nestresovaných buňkách a v odpovědi na DNA poškození je defosforylován. Na C-koncové regulační doméně jsou fosforylovány Ser 315 a Ser 392, acetylovány Lys 320, 373 a 382 a sumoylován je Lys 386, zatímco Ser 376 a 378 jsou fosforylovány v normálních buňkách. V odpovědi na ionizující záření se zvyšuje fosforylace u Ser 6, 9 a 15 v době 30 minut po vystavení stresu.(9) Za nejdůležitější posttranslační modifikace proteinu p53 v souvislosti s poškozením DNA jsou považovány fosforylace na Ser 15 a Ser 392. Fosforylace na Ser 15 představuje časnou odpověď buňky na genotoxický stres.
Dochází k ní aktivací ATM kinázy jak po UV záření, tak po ionizujícím záření a je spojována s indukcí apoptózy.(10) Fosforylace proteinu p53 na Ser 392 zvyšuje asociační konstantu pro tetramerní formu tohoto proteinu a může zvyšovat vazbu specifických sekvencí DNA. Také již zmiňované Chk1 a Chk2 hrají roli i při stabilizaci a následné aktivaci proteinu p53 po poškození DNA, a to jeho fosforylací na serinu 20.
Protein p21 – cyklin dependentní kinázový inhibitor 1A – blokuje aktivitu cyklin dependentních kináz, a tím reguluje buněčný cyklus v G1/S kontrolním bodě. Řídícími molekulami G1 fáze buněčného cyklu jsou cykliny D a cyklin E. Funkce cyklinů D je spojena s Cdk4 a Cdk6, zatímco funkce cyklinu E je spojena s Cdk2. Uvedené komplexy cyklin/cyklin dependentní kinázy fosforylují, a tím inaktivují protein retinoblastomu (Rb), který řídí přechod z G1 do S fáze. Protein p21 tedy zmíněné cyklin dependentní kinázy (Cdk4, Cdk6) inhibuje, v důsledku čehož je Rb protein aktivován defosforylací. Tím dochází k negativní regulaci exprese transkripčního faktoru E2F a přechod z G1 do S fáze je blokován.
Exprese genu p21 je kontrolována proteinem p53. Promotor proteinu p21 obsahuje místo pro vazbu p53, po navázání dochází k aktivaci transkripce. Skrze tuto vazbu tedy protein p21 zprostředkovává p53 dependentní zástavu buněčného cyklu v G1 fázi v odpovědi na různé stresové stimuly.(11) Koncentrace proteinu p21 může být ale regulována i mechanismy, které na p53 nezávisí, kdy p21 může být aktivován například proteinem Ras nebo BRCA-1.(12)
Tumor supresorový protein p16 je klíčovým proteinem pro navození senescence buňky. Vyskytuje se ve třech izoformách, z nichž dvě patří do rodiny inhibitorů kinázy 4 (INK4). Jejich inhibiční účinek je dán schopností tvořit komplexy s Cdk4 a 6, čímž brání vazbě cyklinu D a postupu do S fáze. Třetí izoforma stabilizuje protein p53 tím, že brání jeho degradaci pomocí proteasomu. Navzdory strukturální a funkční odlišnosti všechny izoformy, ať už regulací Cdk4/6 nebo p53, fungují jako kontrola G1 fáze buněčného cyklu.
Apoptóza
Apoptóza je programovaný zánik buněk nenavozující zánětlivou odpověď, jako je tomu u nekrózy. Zda buňka uhyne apoptózou či nekrózou, záleží na několika faktorech. Především je to typ buňky, energetická rovnováha a schopnost syntetizovat nové ATP. Při apoptóze buňka ztrácí nejprve asymetrii fosfolipidů v membráně, dochází ke kondenzaci chromatinu, redukci velikosti jádra a ke štěpení internukleosomové DNA. Poté nastává svraštění buňky, vydouvání membrány a buňka se rozpadá na apoptotická tělíska obklopená zbytky membrány, která jsou v konečné fázi fagocytována.(13)
V apoptotických buňkách je specificky aktivována skupina cystein proteáz, které jsou mezi sebou homologní a jsou součástí velké rodiny proteinů známých jako kaspázy. Kaspázy jsou považovány za hlavní vykonavatele apoptotické cesty, protože zprostředkují většinu změn charakterizujících buněčnou smrt. Jsou syntetizovány jako neaktivní proenzymy tvořené třemi doménami. Signální dráhy vedoucí k apoptóze lze rozdělit na vnější cestu, která vede přes receptory smrti s klíčovým iniciátorem kaspázou 8, a vnitřní cestu, aktivující kaspázu 9, především uvolněním některých faktorů (cytochrom c) z aktivovaných mitochondrií.
Senescence
Některé buňky však po působení genotoxického stresu nepodléhají apoptóze, ale zůstávají permanentně v bloku buněčného cyklu a neproliferují. Po dosažení tzv. Hayflickova limitu buňky nejsou schopny proliferovat a jejich růst je permanentně zablokován. Tento stav je označován jako replikativní senescence a je charakterizován několika znaky, z nichž nejmarkantnějším je zástava buněčného cyklu v G1 fázi, kdy se buňky dále nedělí. Replikativní senescence buněk nastává po zkrácení telomer, speciálních nukleoproteinových struktur, které se nacházejí na koncích eukaryotických chromosomů a jsou tvořeny opakující se sekvencí TTAGGG.
Telomerická oblast reaguje s různými proteiny, které chrání konec chromosomu a zabraňuje spuštění signálních cest, reagujících na poškození DNA. Telomerické sekvence prodlužuje enzym telomeráza, reverzní transkriptáza skládající se z RNA komponenty a katalytické podjednotky, která kompenzuje zkracování telomer během replikace a zabraňuje předčasné senescenci.(8) Kriticky krátké telomery mohou být považovány za určitou formu DNA poškození a označeny fosforylovaným histonem H2AX za vzniku foci.(14) Replikativní senescence je tedy navozena zkracováním telomer po dosažení Hayflickova limitu. Bylo však prokázáno, že senescence může být navozena i předčasně různými vlivy, které způsobují poškození DNA, především její dvojité zlomy (nesouvisí se zkracováním telomer).
Tento stav se nazývá stresem indukovaná předčasná senescence a může být vyvolána UV zářením, ionizujícím zářením, peroxidem vodíku nebo různými chemoterapeutiky, jako jsou doxorubicin, cisplatina a kamptotecin.
Buňky jak v replikativní senescenci, tak v předčasné senescenci mají speciální morfologii, jsou větší, mají rozmanitější škálu morfotypů, menší buněčnou denzitu a s tím spojenou citlivější kontaktní inhibici mezi buňkami. Důležitým znakem buněčné senescence je také zvýšené množství ß-galaktosidázy, což je hydroláza vyskytující se v lyzosomech, kterých je u senenscentních buněk více a zvětšují svůj objem.
Stresem indukovanou předčasnou senescenci můžeme považovat za mechanismus vedoucí k zástavě růstu buněk s rizikem nádorového zvrhnutí. Nádorové buňky totiž exprimují telomerázu a nevstupují do replikativní senescence. Bylo prokázáno, že ionizující záření a různá chemoterapeutika používaná pro léčbu nádorů způsobují stresem indukovanou senescenci jak u normálních buněk, tak také u buněk nádorových. Působení ionizujícího záření nebo chemoterapeutik neovlivňuje délku telomer, způsobuje však poškození DNA, zejména dvojité zlomy, a tím vyvolává předčasnou senescenci.
Vstup do senescence zprostředkovávají dvě základní, výše zmíněné signální cesty – p53/p21 a p16/Rb. Protein p21 inhibuje aktivitu cyklinu Cdk2 a blokuje přechod buněk z G1 do S fáze buněčného cyklu. Výsledkem toho jsou buňky permanentně zastavené v G1 fázi. Tato cesta se zdá být regulována telomerickými signály. Regulační cesta nezávislá na zkracování telomer vede přes protein p16, který inhibuje aktivitu kináz Cdk4 a Cdk6 a indukuje hypofosforylaci Rb proteinu. Tím je znovu inhibován přechod buněk z G1 do S fáze buněčného cyklu.(15)
Kmenové buňky
Kmenové buňky jsou zvláštním typem buněk, které se mohou vyskytovat téměř ve všech typech tkání po celý život. Byly izolovány například z kostní dřeně, mozku, kůže, vlasových folikulů, kosterní svaloviny nebo zubní pulpy. Kmenové buňky charakterizuje řada výjimečných vlastností.
Nejdůležitější z nich je vysoký proliferační potenciál, schopnost nekonečné sebeobnovy (schopnost proliferace přes 50 buněčných populací) a schopnost diferencovat se do široké škály buněčných typů. Hlavní úlohou kmenových buněk je zajišťovat rozvoj tkáně, udržovat tkáňovou homeostázu a opravu tkáně při jejím poškození.
Mezenchymální kmenové buňky jsou nehematopoetické kmenové buňky přítomné v kostní dřeni a schopné diferencovat se do různých typů buněčných linií – osteoblastů, chondrocytů, buněk endotelu a také do buněčných linií podobných neuronům. Do osteogenní linie se mohou diferencovat působením dexametazonu, 10% fetálního telecího séra, ß-glycerol fosfátu a askorbátu. Po jednom týdnu je možné pozorovat akumulaci vápníku a nárůst aktivity alkalické fosfatázy 4–10krát. Chondrogenní diferenciace je pozorována po působení transformujícího růstového faktoru ß3.(16)
Mezenchymální kmenové buňky mohou být využity v systémových transplantacích u celkových onemocnění, lokálních implantací při místním tkáňovém poškození nebo jako nosič genů v genové terapii. Byly izolovány na základě jejich adherence k plastovým povrchům. Pro získání čisté populace mezenchymálních kmenových buněk se používá monoklonální protilátka Stro-1. Mezenchymální kmenové buňky s podobnou biologickou charakteristikou jako buňky z kostní dřeně byly izolovány také z periferní krve, pupečníkové krve, synoviálních membrán a mléčného chrupu.(17)
Pro využití kmenových buněk v klinické praxi je důležitá jejich relativně snadná dostupnost pro izolaci. Takovýmto zdrojem buněk je zubní pulpa. Kmenové buňky zubní pulpy mají podobné vlastnosti jako mezenchymální kmenové buňky kostní dřeně: jsou vysoce proliferativní a mohou expandovat za Hayflickův limit. Velkou výhodou kmenových buněk zubní pulpy v porovnání s jinými kmenovými buňkami je však jejich snadná dostupnost, vysoká interaktivita s biomateriály, což z nich činí potenciální ideální elementy pro rekonstrukci tkáně.
Otázkou je, jak kmenové buňky reagují na genotoxický stres vyvolaný ionizujícím zářením? Hematopoetické kmenové buňky můžeme rozdělit pomocí čtyřbarevné flowcytometrie na buňky dlouhodobě obnovující krvetvorbu (LT-HSC), které odpovídají na ozáření především indukcí senescence, a dále buňky rychle obnovující krvetvorbu (ST-HSC) a buňky progenitorové, které odpovídají na ozáření především indukcí apoptózy. U LT-HSC je vznik senescence spojován s nedostatečnou funkcí těchto buněk ve stáří. Je třeba si uvědomit, že buňky v senescenci se nemohou dělit, a tudíž při další zátěži organismu genotoxickým stresem nemohou obnovit např. poškozenou krvetvorbu.
Byla také prokázána akumulace ?-H2AX foci u LT-HSC izolovaných od starých myší.(18) Mezenchymální kmenové buňky izolované z kostní dřeně neumírají po ozáření apoptózou, ale ztrácejí svou schopnost proliferace. Po ozáření dávkou 2,5–15 Gy dochází ke zkracování telomer, buňky se nedělí, ale ani nejsou zničeny. Zároveň stoupá aktivita galaktosidázy, což je typickým znakem senescence.(19) Také busulfan (alkylační látka způsobující DNA poškození) vyvolává u lidských fibroblastů (linie WI38) senescenci především přes aktivaci extracelulárních signálů aktivujících kinázu Erk a p38 MAPK (mitogen activated protein kinase).
Tato kaskáda je nezávislá na cestě aktivované přes p53.(20) Indukce senescence busulfanem je aktivována reaktivními kyslíkovými radikály. Je-li zabráněno vzniku těchto radikálů např. inkubací s N-acetylcysteinem, snižuje se aktivace Erk a p38 MAPK kináz a také klesá senescence indukovaná busulfanem. Je třeba studovat odpověď kmenových buněk normálních tkání na cytostatické látky a srovnávat tuto odpověď s odpovědí nádorových buněk na látky, které jsou užívány v protinádorové terapii. Ochrana zdravých tkání před indukcí senescence může vést k novým protinádorovým strategiím chránícím normální tkáně před senescencí a indukující u nádorových buněk apoptózu.
Tato práce vznikla za podpory výzkumného záměru MSM 0021620820 a grantu GAČR 304/09/1568.
O autorovi: 1Mgr. Jana Ćmielová, 1PharmDr. Darina Muthná, Ph. D., 2prof. RNDr. Jiřina Vávrová, CSc., 1MUDr. Martina Řezáčová, Ph. D.
1Univerzita Karlova v Praze, Lékařská fakulta Hradec Králové, Ústav lékařské biochemie 2Univerzita obrany, Fakulta vojenského zdravotnictví, Katedra radiobiologie
e-mail: cmielova@lfhk.cuni.cz
