

Téměř před dvaceti lety byl totiž rutinní výzkum cytochromu P450 v počátcích a i reglementy SÚKL (Státní ústav pro kontrolu léčiv) byly méně striktní, než je tomu dnes. Nyní je již jen málo podnětů pro farmaceutické společnosti k provádění výzkumu u léčiv, jejichž patentová ochrana vypršela. V této kapitole chceme sumarizovat data o psychotropních léčivech ze skupin antidepresiv (selektivní inhibitory zpětného vychytávání serotoninu, tricyklická a některá další antidepresiva), anxiolytik, hypnosedativ a antipsychotik.
Summary
Dostálek, M., Turjap, M., Grundmann, M. Drug interactions at the level of biotransformation processes Tricyclic antidepressants, typical antipsychotics, lithium, and benzodiazepines were the mainstay of psychiatric interventions until the last 20 years, when many new drugs become available. Surprisingly little is known about the metabolism of the older drugs. Almost twenty years ago, cytochrome P450 bench techniques were in their infancy, and SUKL (State Institute for Drug Control) was less stringent. Currently, there is little incentive for drug companies to conduct research on older drugs after their patenta have expired. In this chapter, we review the psychotropic drugs in groups: antidepressants (selective serotonin reuptake inhibitors, other commonly used antidepressants, and tricyclics), anxiolytics, hypnotics (benzodiazepines and others) and antipsychotics.
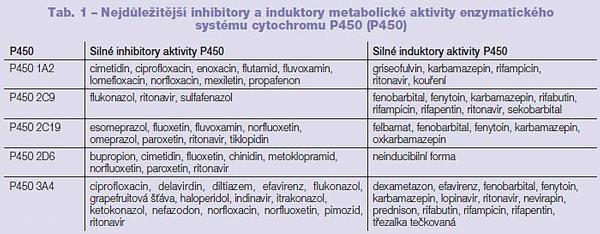
Identifikace interakcí léčiv a jejich kvantifikace dovolují vytvoření strategií pro jejich využití (v případě prospěšných interakcí, například kombinace antihypertenzív), případně omezení jejich výskytu (v případě škodlivých interakcí, například kombinace simvastatinu a itrakonazolu) v klinické praxi. Jejich znalost a popis umožňují například změnu dávkovacích schémat, nahrazení léčiva léčivem s jiným metabolickým profilem a podobně. První informace o lékových interakcích kandidátů nových léčiv jsou často popisovány již v průběhu preklinických experimentálních prací na zvířecích modelech, v mnoha případech se však objevují až při prvním podání léčiva do organismu v průběhu první fáze klinického testování a dále potom po uvedení léčiva na trh. Podle mechanismu vzniku jsou lékové interakce děleny do tří kategorií: 1. lékové interakce na základě interakce s enzymatickým systémem cytochromu P450 a enzymy druhé fáze biotransformace (1. a 2. fáze biotransformace), 2. lékové interakce na základě interakcí s transportními mechanismy v organismu (3. fáze biotransformace), 3. farmakogeneticky podmíněné interakce. Některá léčiva či složky potravy vyvolávají zvýšenou enzymatickou aktivitu, tzv. indukci, jiná léčiva či složky potravy naopak aktivitu enzymů tlumí a způsobují tzv. inhibici (Tab. 1). Protože se nejedná o specifický děj, velmi často se tato změna metabolické aktivity promítá i do biotransformační přeměny jiných látek; hovoříme o indukci, resp. inhibici zkřížené. Důsledkem enzymatické indukce po opakovaném podání látky není jenom zrychlení biotransformace, ale i snížení plazmatických hladin a terapeutického účinku. K této situaci dochází v případech, kdy metabolity léčiva mají malý nebo žádný farmakologický účinek. Pokud však mají metabolity větší farmakologický účinek (dochází k bioaktivaci proléčiv), resp. vyšší toxicitu než mateřská látka, může dojít při enzymatické indukci k projevům intoxikace. Jiná léčiva či složky potravy mohou biotransformační procesy inhibovat. Výsledkem inhibice biotransformačních procesů je většinou prodloužení farmakologického účinku inhibici vyvolávající látky i současně podávaných léčiv, která se eliminují stejnou biotransformační cestou jako látky vyvolávající inhibici. V případě, že se léčiva metabolicky transformují na aktivní meziprodukty, může inhibice biotransformačních procesů farmakologicky snížit účinek v neaktivní formě podávaného léčiva.
Antidepresiva
Antidepresiva jsou léčiva, která jsou užívána k mírnění stavů deprese. Dělí se na 1. antidepresiva první generace (tricyklická a tetracyklická antidepresiva), 2. antidepresiva druhé generace (viloxazin), 3. antidepresiva třetí generace (SSRI – selective serotonin reuptake inhibitors), 4. antidepresiva čtvrté generace (SNRI – serotonin norepinephrine reuptake inhibitors a NDRI – norepinephrine-dobLiéokptrosavynécs pamine reuptake inhibitors), 5. ostatní (NaSSA – noradrenergic and specific serotoninergic antidepressants, SARI – serotonin antagonist reuptake inhibitors, NRI – norepinephrine reuptake inhibitors, SSE – specific serotonin enhancers).
Tricyklická antidepresiva (TCA)
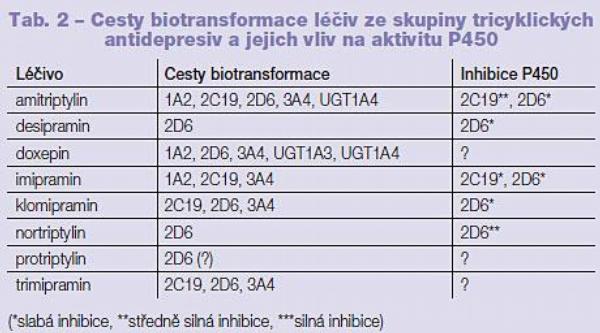
Tricyklická antidepresiva dostala svůj název podle chemické struktury, ve které jsou tři aromatické kruhy (strukturálně blízké fenothiazinu). Léčiva této skupiny byla zprvu používána jako antihistaminika se sedativním účinkem. Amitriptylin a imipramin jsou prototypy klasifikované jako léčiva s inhibičním účinkem na zpětné vychytávání norepinefrinu a serotoninu. Tricyklická antidepresiva mohou být rozdělena do dvou skupin na terciární a sekundární aminy, podle způsobu vazby dusíkatého atomu. Do skupiny terciárních aminů řadíme například amitriptylin, klomipramin, doxepin, imipramin a trimipramin; do skupiny sekundárních aminů potom například desipramin, nortriptylin a protriptylin. Biotransformace této skupiny látek je velmi komplexní, terciární aminy jsou nejprve demetylovány na sekundární aminy (amitriptylin na nortriptylin; imipramin na desipramin; doxepin na desmetyldoxepin; trimipramin na desmetyltrimipramin; klomipramin na desmetylklomipramin) a následně hydroxylovány a vylučovány po konjugaci s kyselinou glukuronovou. Obecně platí, že P450 2D6 je zodpovědný za hydroxylaci sekundárních aminů, některé z dalších P450 jsou zodpovědné za demetylaci. Velmi jedinečný je proces biotransformace nortriptylinu, kde je demetylace zprostředkována P450 2D6 a hydroxylace P450 3A.(1) Právě interakce tricyklických antidepresiv a léčiv ze skupiny SSRI (silných inhibitorů P450 2D6) přiměly ke zkoumání vlivu jednotlivých léčiv užívaných v psychiatrii na aktivitu P450 a vlivu polymorfismu tohoto enzymu na farmakoterapii. Hladina imipraminu je výrazně snížena u kuřáků, protože demetylace P450 1A2 je zde kritickým krokem biotransformační přeměny.
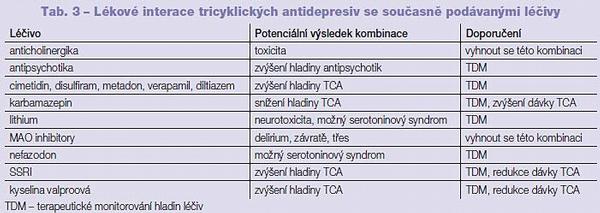
Tricyklická antidepresiva jsou inhibitory P450. Amitriptylin a imipramin jsou inhibitory aktivity P450 2C19 a 2D6 a mohou tak signifikantně zvýšit plazmatickou hladinu fenothiazinových derivátů, plazmatická hladina olanzapinu je imipraminem zvýšena o 20 %; klomipramin, desipramin a nortriptylin jsou inhibitory aktivity P450 2D6. Dosud nejasný je vliv protriptylinu a trimipraminu na aktivitu P450 (Tab. 2, 3, 4, 5).
Selektivní inhibitory zpětného vychytávání serotoninu
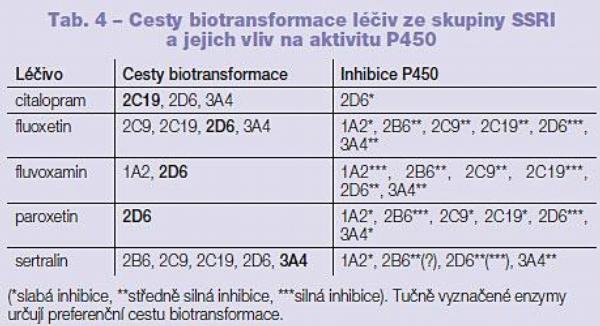
Selektivní inhibitory zpětného vychytávání serotoninu (SSRI, Selective Serotonine Re-uptake Inhibitors) jsou jedny z nejčastěji medikovaných antidepresiv zejména díky bezpečnému profilu a dobré toleranci ze strany pacientů. Každý ze šesti zástupců SSRI má jedinečný biotransformační profil a inhibiční vliv na biotransformační aktivitu enzymatického systému cytochromu P450 (dále jen P450). Podobně jako mateřské látky, tak i jejich aktivní metabolity přispívají k četným lékovým interakcím; například norfluoxetin jako aktivní metabolit fluoxetinu je ve srovnání s mateřskou látkou mnohem silnějším inhibitorem aktivity P450 3A4. Vzhledem ke skutečnosti, že poločas eliminace norfluoxetinu je 7–17 dnů, u fluoxetinu je to pouze 1–3 dny, inhibiční efekt norfluoxetinu na aktivitu P450 přetrvává v organismu i po ukončení terapie fluoxetinem.(2)
Citalopram a S-citalopram (Citalec, Seropram a další)
Citalopram je chirální látka (S-citalopram je co do farmakologického účinku silnějším chirálním enantiomerem citalopramu). Obě látky procházejí při biotransformačních procesech demetylační reakcí na P450 2C19, 2D6 a 3A4. Obě tyto látky jsou inhibitory aktivity P450 2D6,(3) inhibiční účinek obou látek je však poměrně slabý. Vzhledem k diverzitě v metabolických procesech obou látek a k poměrně slabému inhibičnímu účinku na aktivitu P450 2D6 bylo popsáno pouze několik interakcí. Ovšem plazmatické hladiny citalopramu a S-citalopramu mohou být velmi výrazně ovlivněny, pokud je současně podán silný inhibitor aktivity P450 2C19, 2D6 a 3A4. Citalopram neovlivnil při současném podání hladinu klozapinu ani risperidonu.(4)
Fluoxetin (Deprenol, Deprex, Floxet, Prozac a další)
Fluoxetin je chirální látkou, u jehož R-enantiomeru byl v klinických studiích popsán poměrně silný kardiotoxický účinek. Fluoxetin a jeho aktivní metabolit norfluoxetin jsou substráty P450 2C9, 2C19, 2D6 a 3A4.(3) Společně jsou obě látky silnými inhibitory aktivity P450 2D6 a středně silnými inhibitory aktivity P450 2B6, 2C9, 2C19 a 3A4.(3) Vzhledem k zapojení více P450 do biotransformačních procesů fluoxetinu současné podání jiného inhibitoru aktivity P450 většinou nemá na plazmatickou hladinu léčiva v organismu vliv. U pacientů, kteří užívají fluoxetin, je třeba brát v úvahu, že současné podání jiných substrátů P450 2D6 povede k výraznému zvýšení plazmatických hladin těchto léčiv (koncentrace desipraminu byla při současném podání s fluoxetinem zvýšena 4krát). Současné podání fluoxetinu a risperidonu vedlo k 75% plazmatické hladiny risperidonu. Hladina olanzapinu ani quetiapinu nebyla však současným podáním fluoxetinu ovlivněna. Hladina klozapinu byla zvýšena současným podáním fluoxetinu o 70 %.
Fluvoxamin (Fevarin a další)
Fluvoxamin je achirální látka, u které nebyl doposud popsán hlavní metabolit. Jedná se o substrát P450 1A2 a 2D6 a velmi silný inhibitor aktivity P450 1A2 a 2C19, a to i ve velmi nízkých koncentracích, středně silný inhibitor aktivity P450 2B6, 2C9, 3A4 a slabý inhibitor aktivity P450 2D6. Při současném podání s látkami, které mají úzké terapeutické rozmezí, jako jsou teofylin, warfarin, fenytoin a tricyklická antidepresiva, je třeba zvýšené pozornosti. Obecně platí, že je-li v průběhu medikace fluvoxaminem nasazen nový lečivý přípravek, začínáme s nízkou dávkou, kterou pozvolna zvyšujeme. Plazmatická hladina fluvoxaminu může být snížena u kuřáků (díky zvýšené aktivitě P450 1A2 indukované cigaretovým kouřem). Fluvoxamin zvýšil hladinu klozapinu v rozmezí 5 až 10krát. Hladina olanzapinu byla při současném podání zvýšena o 100 %, hladina risperidonu byla ovlivněna jen minimálně (zvýšena o 10 %).
Paroxetin (Seroxat a další)
Paroxetin je v léčivých přípravcích dostupný jako S-enantiomer, doposud nebyl popsán jeho hlavní metabolit. Paroxetin je substrátem P450 2D6, malé množství podaného léčiva je vylučováno i P450 3A4.(5) Jedná se o silný inhibitor aktivity P450 2B6 a 2D6 (díky vysoké afinitě k P450 2D6 je tato biotransformační cesta po podání paroxetinu pro jiná léčiva prakticky uzavřena) a slabší inhibitor aktivity P450 1A2, 2C9, 2C19 a 3A4. Pacienti s medikací přípravků biotransformovaných P450 2B6 a 2D6 jsou vystaveni při současném podání paroxetinu četným lékovým interakcím právě v důsledku inhibice těchto enzymů (například plazmatická hladina desipraminu byla po podání paroxetinu zvýšena o 360 %). Paroxetin zvýšil při současném podání hladinu klozapinu o 40 % a risperidonu o 50 %.
Sertralin (Serlift, Zoloft a další)
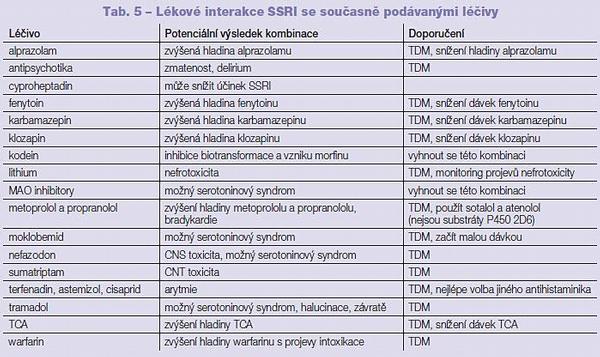
Sertralin je v léčivých přípravcích dostupný jako S-enantiomer, jeho aktivní metabolit desmetylsertralin má stejný inhibiční profil jako mateřská látka.(3) Obě tyto látky jsou středně silnými inhibitory aktivity P450 2D6 a 3A4, což vede ke zvýšené plazmatické hladině léčiv, která jsou zde biotransformována (například fenytoin, warfarin, cyklosporin). Při zvýšené dávce sertralinu dochází k silné inhibici aktivity P450 2D6 (plazmatická hladina desipraminu byla po podání vyšší dávky sertralinu zvýšena o 44 %). In vitro studie popisují silný inhibiční vliv sertralinu na aktivitu P450 2B6, tyto výsledky však nebyly dosud potvrzeny in vivo. (6) Sertralin zvyšoval plazmatickou hladinu diazepamu a zejména pimozidu (díky čemuž byla do příbalového letáku přidána informace o možných interakcích na úrovni P450). Sertralin je znám jako inhibitor glukuronidace (enzymy rodiny UGT), což je nejvýznamnější mechanismus druhé „konjugační“ fáze biotransformace v organismu. Byla popsána intoxikace lamotriginem, který je konjugován právě s UGT. Plazmatická hladina sertralinu může být ovlivněna pouze současným podáním velmi silných induktorů aktivity P450. Sertralin neovlivnil při současném podání hladinu klozapinu, risperidonu ani olanzapinu (Tab. 4).(7)
Inhibitory zpětného vychytávání serotoninu a noradrenalinu, inhibitory zpětného vychytávání noradrenalinu a dopaminu
Venlafaxin (Efectin a další)
Venlafaxin je slabým inhibitorem P450 2D6, biotransformován je P450 2D6 na aktivní metabolit O-desmetylvenlafaxin, který je dále biotransformován P450 2C19 a 3A4.(3) Interakční potenciál této látky však nebyl významně studován. Z dostupných informací byla hladina venlafaxinu zvýšena inhibitory aktivity P450 2D6 (chinidinem, paroxetinem, difenylhydraminem a buspironem). Venlafaxin jako slabý inhibitor aktivity P450 2D6 zvýšil hladinu desipraminu, haloperidolu a imipraminu.
Bupropion (Wellbutrin, Zyban a další)
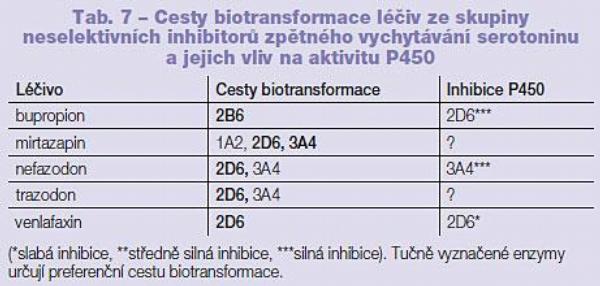
Bupropion je biotransformován P450 2B6, menší zásah do biotransformace bupropionu byl popsán pro P450 1A2, 2A6, 2C9, 2E1, 3A4. Inhibitory aktivity P450 2B6 proto mohou výrazně ovlivinit plazmatickou hladinu bupropionu (ritonavir, efavirenz, nelfinavir, paroxetin, sertralin a další). Plazmatická hladina bupropionu byla snížena současným podáním karbamazepinu. Bupropion jako středně silný inhibitor aktivity P450 2D6 může zvýšit plazmatickou hladinu venlafaxinu, nortriptylinu a desipraminu. Díky nízké afinitě bupropionu k P450 2D6 nejsou popsány změny v hladinách paroxetinu či fluoxetinu při současném podání těchto léčiv (SSRI léčiva jakoby pro bupropion P450 2D6 uzavřela a jeho inhibiční účinek se tak nedostaví).
Další antidepresiva
Mirtazapin (Remeron a další)
Mirtazapin je biotransformován prostřednictvím P450 1A2, 2D6, 3A4 a následně podléhá glukuronidaci. Není popsán inhibiční vliv na aktivitu žádného enzymu ze systému P450. Vzhledem k tomu, že fluvoxamin je inhibitorem všech biotransformačních cest mirtazapinu, je plazmatická hladina mirtazapinu při současném podání zvýšena 4krát. Cimetidin jako jeden z nejsilnějších inhibitorů aktivity P450 1A2, 2C9, 2D6 a 3A4 však zvyšuje plazmatickou hladinu mirtazapinu pouze o 22 %. Možným vysvětlením může být rozdíl v afinitě obou látek k jednotlivým P450.(8) Rifampicin jako silný induktor aktivity P450 3A4 a slabší induktor aktivity P450 1A2, 2C9 a 2C19 signifikantně snižuje plazmatickou hladinu mirtazapinu.
Nefazodon a trazodon (Trittico)
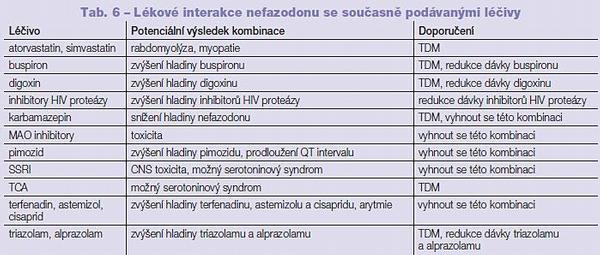
Nefazodon je biotransformován P450 3A4 na tři hlavní metabolity. Jedním z nich je m-chlorofenylpiperazin, který je následně biotransformován P450 2D6.(9) Plazmatická hladina je proto velmi výrazně ovlivněna u polymorfismu P450 2D6 a při současném podání silných inhibitorů aktivity P450 2D6, jako jsou fluoxetin a paroxetin.(9) Plazmatické hladiny obou látek jsou lehce ovlivnitelné i inhibitory a induktory aktivity P450 3A4.(9) Nefazodon je velmi silným inhibitorem aktivity P450 3A4, čímž zvyšuje hladinu současně podávaných látek, jako jsou karbamazepin, simvastatin, cyklosporin, takrolimus, alprazolam a triazolam; kontraindikován je pimozid. M-chlorofenylpiperazin je hlavním produktem biotransformace trazodonu, ten však na rozdíl od nefazodonu není inhibitorem aktivity P450 3A (Tab. 6, 7).
Anxiolytika a hypnotika
Benzodiazepiny
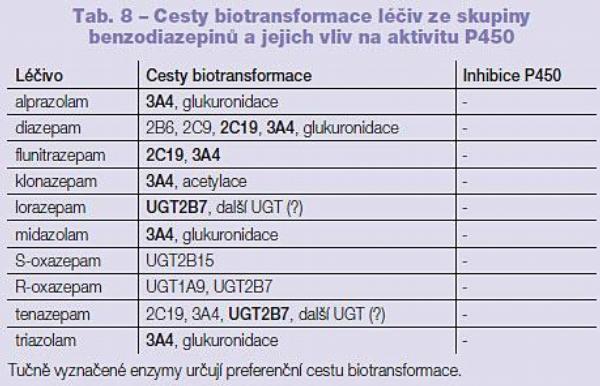
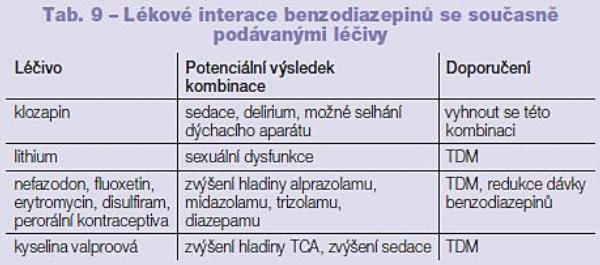
V klinické praxi se můžeme setkat s poměrně velkým množstvím benzodiazepinů. O to překvapivější je skutečnost, že přibližně u jedné poloviny zástupců této skupiny léčiv nebyl proces biotransformace dosud detailně popsán. Mnohé benzodiazepiny jsou biotransformovány P450 3A4 (alprazolam, midazolam, triazolam). Diazepam je biotransformován P450 2C9 a 3A4, minoritní cesty biotransformace diazepamu jsou potom P450 2B6 a 2C9. Flunitrazepam je biotransformován P450 2C19 a 3A4. Všechny tyto benzodiazepiny jsou ve druhé fázi biotransformace glukuronidovány. Klonazepam je částečně biotransformován P450 3A4 a následně je acetylován. Lorazepam a oxazepam jsou biotransformovány primárně glukuronidací (UGT 2B7). Temazepam je eliminován převážně glukuronidací (UGT 2B7, zapojeny jsou však i další UGT) s účasti P450 2C19 a 3A4 (Tab. 8, 9).
Plazmatická hladina benzodiazepinů biotransformovaných P450 3A je ovlivněna současným podáním inhibitorů, případně induktorů aktivity P450 3A (například plazmatická hladina midazolamu je při současném podání flukonazolu zvýšena 4krát). Na druhou stranu itrakonazol jako silný inhibitor aktivity P450 3A4 nezvýšil hladinu temazepamu díky zapojení UGT 2B7 a dalších enzymů rodiny UGT do biotransformace této látky. Současné podání kontraceptiv signifikantně zvýšilo aktivitu zástupců rodiny UGT, a tím i odbourávání lorazepamu, oxazepamu a temazepamu.
Nebenzodiazepiny
Žadný ze zástupců této skupiny léčiv není inhibitorem aktivity P450. Biotransformace všech těchto látek je plně závislá na aktivitě P450 3A4 (Tab. 10).
Buspiron (Anxiron, Buspiron-Egis a další)
Buspiron je biotransformován P450 3A4 na aktivní metabolit 1-pyrimidinylpiperazin. Inhibitory aktivity P450 3A4 tak mohou při současném podání plazmatickou hladinu buspironu zvýšit (nefazodon 20krát, itrakonazol 13krát, erytromycin 5krát, grapefruitový džus 4krát). Induktory aktivity P450 plazmatickou hladinu buspironu naopak snižují, rifampicin snižuje plazmatickou hladinu buspironu až o 85 %. Podobný efekt lze očekávat i u dalších silných induktorů aktivity P450 3A.
Zaleplon (Sonata, Zerene)
Zaleplon podléhá biotransformaci aldehyd dehydrogenázou, částečně je do biotransformace zaleplonu zapojen i P450 3A4. Induktory aktivity P450 však mohou plazmatickou hladinu zaleplonu výrazně snížit, rifampicin snižuje plazmatickou hladinu až o 80 %. Podobný efekt lze očekávat i u současného podání s dalšími silnými induktory aktivity P450. Vliv inhibitorů aktivity P450 3A4 na hladinu zaleplonu je nižší (erytromycin zvýšil plazmatickou hladinu zaleplonu o 34 %). Cimetidin jako silný inhibitor jak aldehyd dehydrogenázy, tak i aktivity P450 3A4 zvýšil hladinu zaleplonu o 85 %.
Zolpidem (Hypnogen, Stilnox)
Zolpidem je biotransformován P450 3A4, 1A2 a 2C9. Hladina zolpidemu je vlivem silných inhibitorů aktivity P450 3A4 zvýšena (ritonavirem o 70 %, ketokonazolem o 60 % a itrakonazolem o 35 %). Tyto interakce jsou však mnohem méně závažné, než tomu bylo u buspironu, a to zejména díky zapojení P450 1A2 a 2C9 do biotransformačních procesů zolpidemu. Hladina zolpidemu je snížena podáním induktoru P450 3A4 (rifampicinem o 58 %).
Zopiklon (Zopitin)
Zopiklon je chirální látka a výsledkem biotransformace je vznik aktivního metabolitu. Do biotransformace zopiklonu jsou zapojeny P450 2C8 a 3A4. Vylučování látky je velmi složité a děje se ledvinami, plícemi a játry (o zapojení jednotlivých orgánů do procesu exkrece zopiklonu zatím není dostatek informací).(10) Silní inhibitoři aktivity P450 3A4 signifikantně zvyšují hladinu zopiklonu (itrakonazol o 77 %). Rifampicin jako silný induktor aktivity P450 hladinu zopiklonu snížil o 66 %.

Antipsychotika
Termíny antipsychotika a neuroleptika zahrnují skupinu léčiv nacházející uplatnění zejména ve farmakoterapii schizofrenie. V moderní medicíně jsou tato léčiva užívána více než padesát let. Reserpin a chlorpromazin byly prvními léčivy této skupiny, která byla používána v terapii schizofrenie. Tato léčiva však byla postupně nahrazena skupinou nových antipsychotik, která vytlačují z terapeutické praxe původní zástupce.
Aripiprazol (Abilify)
Jedná se o chinolonový derivát popsaný v roce 2002. Dehydroaripiprazol je aktivním metabolitem aripiprazolu a je výsledkem biotransformace P450 2D6 a 3A4. Samozřejmě inhibitory aktivity P450 2D6 a 3A4, stejně jako induktory aktivity P450 3A4, budou hladinu léčiva v organismu ovlivňovat. Zatím je však stále málo známo o interakcích a o ovlivnění hladiny aripiprazolu právě kvůli současnému podání látek ovlivňujících aktivitu P450.
Haloperidol (Apo-Haloperidol, Haloperidol-Richter)
Haloperidol podstupuje poměrně složitou biotransformaci, hlavní cestou je redukce, N-dealkylace a hydroxylace za účasti P450 1A2, 2D6 a 3A4. Haloperidol byl popsán jako silný inhibitor aktivity P450 2D6. Inhibitory aktivity P450 2D6 hladinu haloperidolu zvýšily (bupropion, fluoxetin, fluvoxamin), naopak induktory P450 snížily sérovou hladinu haloperidolu (karbamazepin, fenytoin, rifampicin, kouření).
Klozapin (Leponex a další)
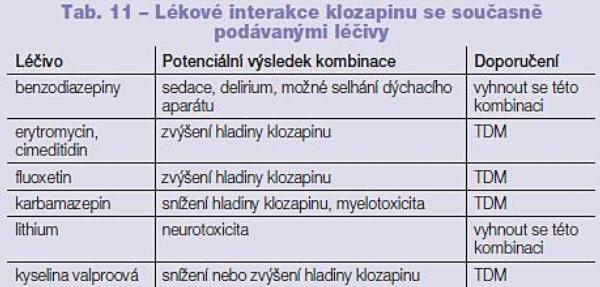
Klozapin je primárně biotransformován na aktivní metabolit norklozapin. Děje se tak prostřednictvím P450 1A2, 2C9, 2C19, 2D6 a 3A4. Klozapin je též biotransformován na klozapin N-oxid, inaktivní metabolit, prostřednictvím P450 3A4 a pravděpodobně i flavin monooxygenázou typu 3.(11) Látky jsou následně konjugovány s kyselinou glukuronovou prostřednictvím UGT 1A4, 1A3 a vyloučena není ani účast dalších forem rodiny UGT. Nejčastější interakce klozapinu jsou popsány s fluvoxaminem (Tab. 11). Tato interakce je však očekávaná; fluvoxamin je, jak bylo uvedeno, silný inhibitor aktivity P450 1A2 a 2C19 a dále inhibitor aktivity P450 2B6, 2C9, 2D6 a 3A4, čímž jsou blokovány všechny cesty biotransformace klozapinu. Interakce byly dále popsány s kontraceptivy, kofeinem a ciprofloxacinem, paroxetinem a fluoxetinem. Velmi matoucí je proto neovlivnění hladiny klozapinu při současném podání jiných inhibitorů aktivity P450, jako jsou ketokonazol, itrakonazol, nefazodon. Vysvětlení najdeme v afinitě léčiv k jednotlivým P450, a proto i interakce s inhibitory aktivity P450 3A jsou zanedbatelné. Silné induktory aktivity P450, jako jsou fenobarbital, karbamazepin, rifampicin a fenytoin, snižují hladinu klozapinu. Kouření hladinu klozapinu v organismu snižuje. Zapomenout nesmíme ani na skutečnost, že klozapin je středně silným inhibitorem aktivity P450 2D6, a proto může dojít k ovlivnění současně podávaných látek biotransformovaných právě P450 2D6 jako v případě nortriptylinu.(12)
Olanzapin (Zyprexa a další)
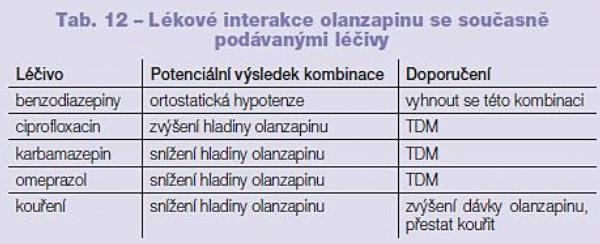
Olanzapin nemá na aktivitu P450 inhibiční ani indukční vliv. Kvantitativně nejdůležitější cestou biotransformace je glukuronidace, popsáno je zapojení UGT 1A4 (probenecid – jako inhibitor aktivity UGT 1A4 – biotransformaci olanzapinu signifikantně snížil). Na druhou stranu karbamazepin, induktor aktivity UGT 1A4, biotransformaci olanzapinu signifikantně zvýšil. Druhou velmi důležitou cestou biotransformace olanzapinu je P450 1A2 (silní inhibitoři aktivity P450 1A2 proto biotransformaci olanzapinu snižují a zvyšují tak jeho hladinu v organismu (ciprofloxacin, kofein, fluvoxamin). Minoritní cestou biotransformace je P450 2D6, fluoxetin zvyšuje hladinu olanzapinu v organismu o 30 % (Tab. 12).
Perfenazin (Perfenazain a další)
Perfenazin byl první léčivo ze skupiny antipsychotik, u kterého byl prokazán vliv polymorfismu P450 2D6 na jeho sérovou hladinu. Hladina perfenazinu byla u PM až 5krát menší v porovnání s EM, po jednorázovém podání 6 mg léčiva. Hlavní cestou biotransformace je dealkylace P450 1A2, 2C19, 2D6 a 3A4. Perfenazin je silným inhibitorem aktivity P450 2D6 a slabým inhibitorem aktivity P450 1A2.
Quetiapin (Seroquel a další)
Quetiapin je biotransformován P450 3A4 a jeho hladina je tak ovlivněna všemi inhibitory tohoto enzymu (ketokonazol zvyšuje hladinu quetiapinu o 300 %, podobný efekt byl popsán i pro erytromycin, klaritromycin, diltiazem, nefazodon). Podobně všechny induktory aktivity P450 3A4 budou hladinu quetiapinu snižovat (fenytoin a karbamazepin). Dosud neznámým mechanismem dochází ke snížení hladiny quetiapinu i při současném podání s thioridazinem.(13)
Risperidon (Rispen, Risperdal a další)
Risperidon, jako středně silný inhibitor aktivity P450 2D6, je biotransformován P450 3A4 a 2D6 na hlavní metabolit 9-hydroxyrisperidon. V literatuře scházejí informace o možných interakcích se silnými induktory či inhibitory aktivity P450. Možným vysvětlením je skutečnost, že metabolit, 9-hydroxyrisperidon, má totožné farmakologické vlastnosti jako mateřská látka.(14) Na druhou stranu plazmatickou hladinu 9-hydroxyrisperidonu byla zvýšena o 45 % při současném podání paroxetinu. Induktory aktivity P450 3A4 snižují koncentraci risperidonu až do neúčinných hladin. Plazmatická hladina karbamazepinu je signifikantně zvýšena současným podáním s risperidonem, mechanismus není zatím popsán a nevylučuje ani možnost ovlivnění aktivity P450 (Tab. 13).(15)
Ziprasidon (Zeldox a další)
Hlavní biotransformační cestou ziprasidonu je aldehydoxydáza (tedy podobně jako u zaleplonu, citalopramu, acikloviru a dalších). Přibližně 30 % podaného ziprasidonu je biotransformováno P450, zejména prostřednictvím P450 3A. V menší míře do biotransformace ziprasidonu zasahuje P450 1A2. Ketokonazol zvyšuje hladinu ziprasidonu o 40 %, v případě karbamazepinu je to o 35 %. Tyto interakce mohou být velmi závažné z důvodu prodloužení QTc intervalu ze strany ziprasidonu (popsáno při současném podání s mesoridazinem, thioridazinem, pimozidem, roperidolem a celou skupinou Ia a III antiarytmik).

Genetická variabilita ve farmakokinetických procesech léčiv
Nejčastější příčinou genetické variability ve farmakokinetických procesech léčiv jsou mutace lokalizované na typických místech genů enzymů, označují se jako polymorfismy. Polymorfismy můžeme v populaci pozorovat na nejrůznějších úrovních, a to nejen na úrovni sekvence DNA či RNA, ale také na úrovních sekvence aminokyselin. Přítomnost polymorfismu ve struktuře DNA se projevuje změnou enzymové aktivity. Enzymový defekt projevující se změnou v metabolické aktivitě je v populaci přenášen autosomálně recesivně, projevy jsou proto nejvýraznější u homozygotů pro defektní gen, mluvíme o pomalých metabolizátorech (PM); výsledná enzymatická aktivita je u nich velmi nízká nebo žádná. Naopak duplikace nebo amplifikace funkčního genu se klinicky projeví urychlením metabolismu, v tomto případě mluvíme o ultrarychlých metabolizátorech (UM). Většinu populace tvoří homozygoti pro normální gen s dostatečnou metabolickou funkcí, mluvíme o rychlých metabolizátorech (EM), což je považováno za jakousi normu v populaci. Z této skupiny se vyčlenila podskupina heterozygotů s jednou defektní a jednou fyziologickou alelou v genomu, normální gen u těchto jedinců zabezpečuje dostatečnou metabolickou aktivitu, ta však nedosahuje úrovně rychlých metabolizátorů a zástupce této skupiny nazýváme intermediární metabolizátoři (IM). Jedinci s fenotypem pomalých metabolizátorů vykazují po podání per os vysoké plazmatické hladiny podaného léčiva a nízké plazmatické hladiny metabolitů podaného léčiva (v důsledku nízkého efektu prvního průchodu léčiva). Rozhodující je, zda sledovaný enzym u podaného léčiva zprostředkuje proces bioaktivace či biodegradace – reakce organismu na podané léčivo je hypoergní, případně hyperergní. Jedinci s fenotypem ultrarychlých metabolizátorů mají podstatně nižší plazmatické hladiny léčiv. Existují dvě základní metody objektivizace aktivity enzymů podílejících se na biotransformaci léčiv. Stanovení fenotypu, tedy aktuální aktivity enzymu, se provádí pomocí standardně užívaných testovacích látek, jež jsou substráty daného enzymu. Podáním testovací látky je podle jejích farmakokinetických vlastností zjišťován poměr mezi koncentrací mateřské látky a příslušného metabolitu. Aktivita enzymu může být stanovena i nepřímo, prostřednictvím detekce mutací v genu sledovaného enzymu, stanovením genotypu. Genotyp jedince zůstává po celý život neměnný. Fenotyp, tzn. aktivita daného enzymu, osciluje a je závislá i na četných negenetických faktorech (věk, pohlaví, strava a další).
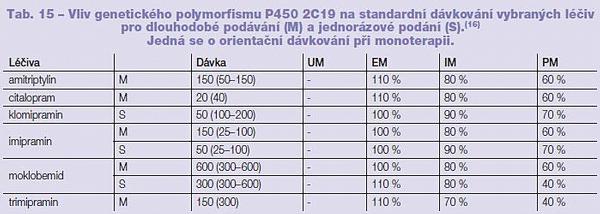
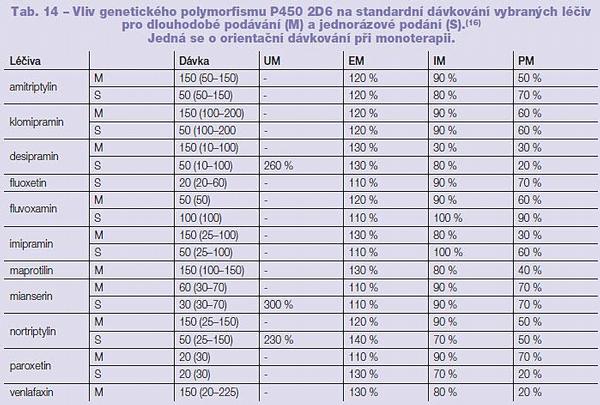
Genetický polymorfismus ovlivňuje první (P450), druhou (konjugační enzymy) i třetí (transmembránové přenašeče) fázi biotransformace. Mezi nejvíce prostudované polymorfismy biotransformačních procesů počítáme polymorfismy P450 1A2, 2C9, 2C19, 2E1 a 2D6, enzymů N-acetyltransferázy a UGT, ze třetí fáze biotransformace je to potom P glykoprotein. Pro příklad důležitosti sledování změn v aktivitě enzymů vlivem polymorfismu uvádíme změny v dávkování vybraných léčiv užívaných v psychiatrické praxi na základě příslušnosti jedince k danému fenotypu (Tab. 14, 15). Vliv polymorfismu na sérovou hladinu lečiv byl popsán například u doxepinu, fluvoxaminu, haloperidolu, imipraminu, klozapinu, mirtazapinu, olanzapinu (substráty P450 1A2); amitriptylinu, citalopramu, imipraminu, klomipraminu, moklobemidu, thioridazinu a trimipraminu (substráty P450 2C19); amitriptylinu, haloperidolu, klomipraminu, desipraminu, fluoxetinu, fluvoxaminu, imipraminu, maprotilinu, mianserinu, nortriptylinu, paroxetinu, perfenazinu, risperidonu a venlaxaxinu (substráty P450 2D6).
Závěrem bychom rádi zdůraznili, že tato práce nebyla podpořena žádnou farmaceutickou společností či distributorem léčiv a data v ní obsažená nemají sloužit k propagaci konkrétních léčiv či léčivých přípravků. Pro ilustraci pokroků v oblasti výzkumu a vývoje nových léčiv jsou uvedena i některá dnes již neregistrovaná léčiva.
Poděkování patří Michaele Geistové za pomoc s přípravou této publikace.
O autorovi: 1PharmDr. Miroslav Dostálek, Ph. D., 1Mgr. Miroslav Turjap, 1, 2doc. MUDr. Milan Grundmann, CSc.
1 Fakultní nemocnice Ostrava, Ústav klinické farmakologie
2 Ostravská univerzita v Ostravě, Fakulta zdravotnických studií
email: dostalekm@hotmail.com
